
CT and MRI diagnosis of middle interhemispheric variant of holoprosencephaly with frontal midline osteoma: A Clinical Case Analysis
-
摘要:
半球中央变异型前脑无裂畸形(MIH)属于前脑无裂畸形的一种变异类型。本文报道1例MIH患者,15岁,男性,出生后即发现前额部凸起,因额部病变影响外观入院手术,入院后的CT和MRI显示额骨正中病变,并偶然发现MIH,伴双侧额叶多小脑回畸形。额骨正中病灶术后病理为骨瘤。本文回顾性分析该例MIH伴发额骨骨瘤的CT和MRI影像特征,并复习国内外文献,总结MIH的影像特征,以提高对该罕见疾病的认识。
Abstract:The middle interhemispheric variant of holoprosencephaly (MIH) is a variant type of holoprosencephaly. In this article, we report the case of a 15-year-old male patient with MIH who presented with a forehead bulge immediately after birth. He was admitted to the hospital for surgery due to a frontal lesion affecting his appearance. CT and MRI revealed a frontal bone lesion and incidentally detected MIH associated with bilateral frontal lobe polymicrogyria. Postoperative pathology of the median frontal bone lesion confirmed it to be an osteoma. This article retrospectively analyzes the CT and MRI imaging features of MIH with frontal midline osteoma and reviews the literature to summarize the imaging characteristics of MIH, aiming to improve the understanding of this rare disease.
-
Keywords:
- CT /
- magnetic resonance imaging /
- holoprosencephaly /
- middle interhemispheric variant /
- osteoma
-
半球中央变异型前脑无裂畸形(middle interhemispheric variant of holoprosencephaly,MIH),又称端脑融合畸形(syntelencephaly)[1]或半球间中央融合型(Middle interhemispheric fusion,MIF) 前脑无裂畸形[2]。MIH属于前脑无裂畸形的一种变异类型,为少见的先天性前脑发育畸形,由1993年Barkovich等[3]首次报道。由于该疾病罕见,影像科医师可能仅诊断胼胝体发育不良。
国内外文献MIH伴额骨正中骨瘤病例尚未见报道,现报道1例15岁MIH伴额骨正中骨瘤的男性患者,总结该疾病的CT和MRI特征,以提高影像和临床医师对该病的认识。
1. 病历资料
1.1 临床资料
患者,男,15岁,出生后即发现前额部凸起,后逐渐生长变大,无压痛,无触痛,无头痛,无恶心呕吐,无肢体无力,随年龄逐渐生长,外院头颅CT诊断巨脑回,胼胝体发育不良,额骨异常发育。患者就读当地初中三年级,由于容貌外观的原因入院拟行额骨病灶切除术。
体检:前额部正中局部隆起,3 cm×4 cm左右,触之较硬,无压痛,局部皮肤毛发生长(图1(a))。患者生长发育未见明显异常,谈吐未见明显异常。
家族史:其母亲怀孕前有糖尿病史,具体用药情况不详。
1.2 影像学表现
术前行头颅CT平扫(图1 (b)~(f))。容积再现成像(图1(b))显示患者额部正中隆起。矢状位骨窗(图1(c))显示前额部正中的额骨外板向前隆起,顶部正中的额骨内板向颅内隆起。轴位脑窗(图1(d))显示额叶后部局部融合(红色箭头),双侧外侧裂于顶部相连(黄色箭头);双侧额叶前部见呈多小脑回畸形改变的深折的不规则增厚皮层(蓝色箭头)。矢状位脑窗(图1(e))显示胼胝体体部(红色箭头)、嘴部缺如,胼胝体膝部和部分胼胝体压部存在(蓝色箭头);轴位脑窗(图1(f))显示透明隔缺失。术后CT平扫容积再现成像(图1(g))显示患者额骨隆起区域术后平坦,钛板塑性贴合。
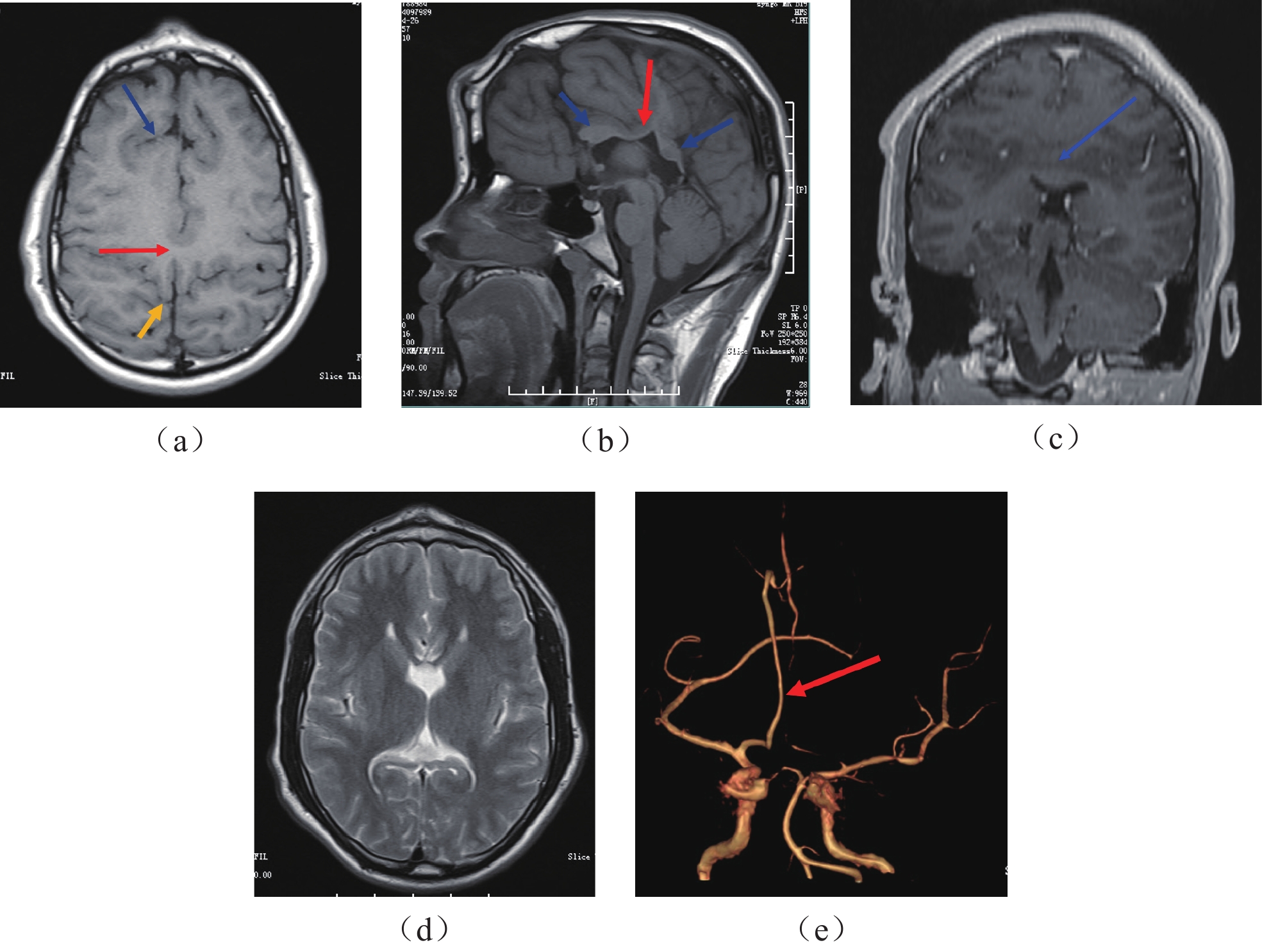
术前头颅MRI平扫轴位T1 WI(图2 (a))显示额叶后部局部融合(红色箭头),双侧外侧裂垂直走向于顶部相连(黄色箭头),双侧额叶前部见深折的不规则增厚皮层(蓝色箭头),呈多小脑回畸形改变;MRI平扫正中矢状位T1 WI(图2(b))显示胼胝体体部(红色箭头)、嘴部缺如,胼胝体膝部和部分胼胝体压部存在(蓝色箭头);前额部的额骨外板向前隆起,局部板障增厚,顶部额骨颅骨内板向颅内隆起,局部板障增厚;MRI平扫额叶后部冠状位T1 WI(图2(c))显示双侧额叶后部局部融合,双侧大脑半球的脑白质相连(蓝色箭头); MRI平扫轴位T2 WI(图2(d))显示透明隔缺失;MRA(图2(e))显示单根大脑前动脉(箭头)。
1.3 手术与病理
全麻下神经导航下行额部颅骨病损切除和颅骨修补术,靠近病变的异常骨质血供非常丰富,松质骨有较多的血喷出,将后方内生型骨质异常逐步切除,随后导航指引下完整铣下外生型骨质病变,右侧额窦似有轻微打开,额窦粘膜完整,额窦表面颅骨较厚,大于0.5 cm,取钛板,塑型满意,钛钉固定妥当。
术后病理:(颅骨病变)骨组织两块,大者大小5.3 cm×4.5 cm×2 cm,小者大小4.5 cm×3.2 cm×1.9 cm,质硬。免疫组化结果:4号蜡块:S-100(+),MDM2(−),CDK4(−),CD34(−),CD117(−),CD43(−),CD10(−)。病理诊断:(颅骨病变)符合骨瘤。
1.4 随访
患者额骨中线骨瘤术后外观明显改善,MIH和双侧额叶多小脑回畸形目前临床无症状,需进一步随访了解有无随年龄显示的上述颅脑先天畸形导致的临床症状(如癫痫等)。
2. 讨论
前脑无裂畸形(holoprosencephaly,HPE),又称全前脑畸形,是一种前脑不能完全分裂成左右半球的前脑畸形[4]。对中国2007年-2014年超过一千万新生儿中前脑无裂畸形的一项研究观察到中国的 HPE 患病率呈上升趋势[5]。HPE患病率于活产儿中约1/
16000 ,胚胎中高达1/250,仅1/10000 婴儿可存活超过1年[4]。1963年,DeMyer提出了经典的HPE分类,根据前脑分裂的程度分为三型:①无脑叶型,大脑半球完全不分离,伴单个脑室;②半脑叶型,仅前部脑叶未能分离,但顶枕区被半球间裂和大脑镰分开;③脑叶型,仅额叶的最下部分融合[6]。随着研究的深入陆续增加了以下几种HPE类型:①半球中央变异型 (middle interhemispheric variant of holoprosencephaly,MIH):MIH是HPE的一种亚型,由1993年Barkovich等[3]发现3名患者前额叶区域存在半球间裂,而额叶后部和顶叶区域的半球未分离,首次提出MIH为HPE的一种新的变异类型; ②milder、minimal型和微型(microforms)HPE,分别为视隔发育不良、视前区不分离以及大脑发育正常而仅面部异常特征(眼距过近和上颌正中孤立中切牙)[6]。
MIH约占HPE的2%~17%,无已知的种族或性别偏好[1, 7]。MIH患者通常不能观察到严重的中线颅面异常[8]。
2.1 病因及发病机制
HPE的病因包括染色体异常、孟德尔综合症疾病、环境因素(巨细胞病毒感染、产前接触药物、孕产妇糖尿病和酗酒等)和多个HPE相关基因的杂合变异。母亲糖尿病(妊娠期和妊娠前)是最明确的HPE危险因素。糖尿病母亲的婴儿中HPE的患病率为1%至2%。母亲接触酒精是另一个已知的危险因素[5, 6, 9]。
MIH畸形与经典HPE有很大不同,MIH的基底前脑相对保留,前部半球间裂形成良好,丘脑受累程度大于尾状核和下丘脑,胼胝体受累位置不同,眼距和视交叉形成大多数正常,这些表明MIH具有与经典HPE不同的胚胎发生机制[10]。MIH是由于神经管闭合后胚胎顶板的有丝分裂细胞凋亡异常,导致半球间裂形成障碍所形成的双侧大脑半球融合畸形,与位于13 q染色体的ZIC2基因异常有关[11]。
2.2 临床特征
不同类型的HPE临床表现差异较多,各种类型的HPE的预后各不相同,主要取决于严重程度和相关并发症。无脑叶HPE或患严重面部畸形的患儿很少在新生儿期存活,而面部异常不太严重的儿童则可存活数月。仅少数HPE患儿能够存活到成年[5]。Weiss等[9]研究20例15岁及以上的前脑无裂畸形的青少年和成人群体(其中25%为MIH),发现不同的 HPE 亚型个体均可存活到成年,随着前脑无裂畸形患者年龄的增长,临床上需关注与痉挛相关的并发症和儿童时期可能不存在的精神和眼科异常。
MIH型HPE患者临床表现在HPE谱系中相对轻微,大多数 MIH型HPE患者有轻度至中度的面部畸形,包括眼距过宽、唇裂和腭裂,常于产前或儿童早期被诊断。最常见的临床发现为癫痫发作、运动功能障碍(最常见轻度至中度痉挛,其次肌张力减退)、智力发育迟缓,患者通常存在言语和口腔运动发育迟缓。但通常不出现其经典的HPE常见的内分泌功能障碍、舞蹈手足徐动症或严重的中线颅面异常[12-13]。MIH也可能无任何症状,Özdemir等[12]报道一例42岁无症状(无神经系统疾病和认知障碍/面部畸形)女性,因中度头痛数周影像检查偶然发现的MIH患者。
2.3 影像学表现
MIH型HPE的CT和MRI特征性表现如下:①双侧大脑半球前额叶和枕叶半球纵裂及大脑镰形成,而额叶后部和/或顶叶融合[11];②双侧外侧裂呈垂直方向,且通常双侧外侧裂在顶部相连,导致顶部区域大脑轴位可见异常横裂。外侧裂的这种异常形态有助于与其他经典HPE亚型区分,无脑叶型HPE的外侧裂通常不存在,半叶型和脑叶型HPE的外侧裂较宽且向前移位[13];③ MIH的胼胝体畸形表现为胼胝体的体部畸形程度最严重,而膝部和压部受累相对较轻[11];④ CTA或MRA显示单根大脑前动脉。MIH型和经典的HPE均可见前动脉循环异常(单根大脑前动脉)[13];⑤弥散张量成像(diffusion tensor imaging,DTI)可比传统MRI更清楚揭示MIH型的异常白质改变[14]。
由于胼胝体畸形相对多见,大部分影像医生容易发现胼胝体的异常。胼胝体从前到后由四部分组成:嘴部、膝部、体部和压部。胼胝体的正常发育顺序为从头到尾,依次是胼胝体膝后部、胼胝体的体前部、胼胝体膝部前部同时伴胼胝体的体后部、压部和嘴部[2, 15]。Coll等[2]研究提出MIH存在不典型和罕见的胼胝体发育不全,即胼胝体膝部和压部发育正常,但胼胝体的体部部分缺失。与正常胚胎胼胝体前后方向的发生相反,MIH是目前已知的唯一的胼胝体前或中部未形成的情况下却形成胼胝体后部的疾病,这有助于将其与其他HPE形式区分开来[14]。
产前检查主要应用超声,但超声对轻型的HPE准确率较低,胎儿MRI为准确诊断HPE和确定亚型的首选方法[4]。
HPE通常与各种中线结构异常相关,包括静脉窦、动脉、静脉和脑膜血管畸形,颅外骨骼缺陷在HPE中很常见,包括脊柱侧凸、肋骨和/或椎骨异常、桡骨发育不良、马蹄足、拇指缺如、多指畸形、短指畸形、先天性趾侧弯、先天性指屈曲和短掌骨畸形等[16]。Fujino等[1]首次报道1例MIH伴发颅骨膜血窦。
骨瘤是生长缓慢的良性病变。其特征是致密的板状骨或松质骨增殖和沉积。通常发生在颅面部区域。颅骨骨瘤是一种生长缓慢的良性骨肿瘤, 无侵袭性,可发生于任何年龄,可发生于颅骨任何部位。熊南翔等[17]将颅骨骨瘤分为4型:①颅底骨瘤:最多见;②颅盖骨瘤:分颅内生长和颅外生长两种;③硬脑膜骨瘤:大多位于大脑镰旁;④脑实质内骨瘤:极少见。骨瘤的发病机制尚未完全阐明,可能由于AP1易感基因突变、感染和创伤[18-19]。
国内外文献中未见MIH伴发额骨正中骨瘤的病例。本文病例额骨正中骨瘤出生即发现,额部隆起区存在毛发,推测与形成MIH型HPE的胚胎期原因有关。该额骨正中骨瘤外观仅表现为额部隆起,影响外观,CT同时发现其额骨内板区域向颅内隆起,随着骨瘤的增大可能会压迫局部脑实质。另外该MIH患者除了典型的MIH的影像表现,同时伴发双侧额叶多小脑回畸形,国外文献报道MIH可伴发多小脑回畸形[20]。
2.4 治疗与预后
MIH患者出现最常见的临床(癫痫发作、运动功能障碍和智力发育迟缓、言语和口腔运动发育延迟)时需要对症治疗。MIH患者可伴发其他的先天畸形,需要根据不同畸形的临床表现对症治疗。
前脑无裂畸形的遗传学诊断较重要,包括产前遗传学诊断和产后遗传学诊断。①产前遗传学诊断:对于家族史阳性、父母疑似患HPE、产前胎儿超声和/或MRI结果异常的高危孕妇,建议妊娠期行家系分析、细胞遗传学分析和基因检测;②产后遗传学诊断:许多轻型HPE病例是在出生后的婴儿期和儿童期才确诊。对可疑的HPE患儿推荐行脑部MRI检查,此外可对患儿行遗传学分析。对死产或终止妊娠时,应考虑从死胎身上获取DNA样本,以便将来进行基因研究,未来怀孕的复发风险取决于遗传异常的类型,如果父母一方携带的突变具有不完全外显率,则复发风险高达50%,如果是非整倍体,如13号三体,复发风险则为1%[4]。
本例患者临床上未表现出常见的MIH临床症状,仅由于伴发额骨中线骨瘤入院手术偶然发现。该患者双侧额叶存在多小脑回畸形,尚未出现明显临床征象,但需要定期随访。颅面骨瘤病变常无症状,无症状骨瘤癌变的风险较低,通常不需要立即切除,但对生长快、影响面容且有症状的骨瘤应手术切除。颅面骨瘤预后良好,复发率低。但骨瘤发生在儿童和青少年时需排除Gardner综合征,Gardner综合征又称家族性多发性结肠息肉-骨瘤-软组织瘤综合征,是家族性腺瘤性息肉病一种亚型(常染色体显性遗传病变)。骨瘤切除后需要长期随访[19]。
2.5 鉴别诊断
MIH的诊断主要依据CT和MRI,其影像学表现较有特征,需要注意观察是否伴发其他的颅脑先天畸形。MIH的鉴别诊断主要如下:①经典的脑叶型前脑无裂畸形:仅额叶的最下部分融合,而额叶后部和顶叶中线区域无融合[6];②视隔发育不全:透明隔缺失或形成不完全,但双侧大脑半球分离良好,胼胝体正常[6];③双侧脑裂畸形:双侧大脑半球见横行裂隙,从脑表面沿伸至室管膜下,裂隙两侧衬不规则增厚的灰质层,其表面有软脑膜与室管膜相连,双侧的大脑半球裂隙与脑室相通,而双侧大脑半球分离良好,无中线区域脑叶融合[21];④双侧大脑外侧裂周围多小脑回畸形:双侧外侧裂区存在异常向内折叠的增厚皮层,而双侧大脑半球和脑室分离良好,无中线区域脑叶融合[20]; ⑤额骨内板增生症:该病是一种累及额骨内表面骨形成的特发性疾病,影像学典型表现为双侧额骨内板对称增厚,且不累及额骨中线区域[22],该病无颅脑先天畸形表现。
3. 结论
总之,MIH罕见,临床症状较轻的患者往往由于行头颅CT或MRI检查时偶然发现。本病例出生时发现额部正中病变,但未出现MIH常见的智力异常、癫痫等症状,本病例由于额骨正中病变影响容貌入院手术的术前影像学检查偶然发现MIH。影像科医师认识该病的特征表现有助于及时诊断MIH,当头颅 CT和MRI发现不典型胼胝体发育不良(胼胝体的体部畸形程度严重,而膝部和压部表现正常或受累相对较轻)时,需仔细观察大脑半球于额叶后部和/或顶叶是否融合,当CTA或MRA发现单根大脑前动脉时也需要考虑MIH的可能。由于MIH涉及多个系统,一旦诊断MIH,需要多学科协作诊治,对患者遗传学的诊断有助于早期诊断和更及时的诊治,孕妇需避免接触致畸的多种因素 [4],患糖尿病的母亲在妊娠前和妊娠期更需要关注胎儿遗传学的诊断。
-
[1] FUJINO S, ENOKIZONO M, IHARA S, et al. Sinus pericranii associated with syntelencephaly: A case report[J]. BMC Neurology, 2022, 22(1): 316. DOI: 10.1186/s12883-022-02764-5.
[2] COLL MASFARRÉ S, MAJÓS TORRÓ C, AGUILERA GRIJALVO C, et al. Middle interhemispheric fusion[J]. European Radiology, 1998, 8(4): 631-633. DOI: 10.1007/s003300050450.
[3] BARKOVICH A J, QUINT D J. Middle interhemispheric fusion: An unusual variant of holoprosencephaly[J]. American Journal of Neuroradiology, 1993, 14(2): 431-440.
[4] 王志燕, 宋涛. 前脑无裂畸形诊断与治疗[J]. 中华整形外科杂志, 2024, 40(3): 325-330. DOI: 10.3760/cma.j.cn114453-20231028-00148. WANG Z Y, SONG T. Diagnosis and treatment in holoprosencephaly[J]. Chinese Journal of Plastic Surgery, 2024, 40(3): 325-330. DOI: 10.3760/cma.j.cn114453-20231028-00148.
[5] YI L, LIU Z, DENG C, et al. Epidemiological characteristics of holoprosencephaly in China, 2007-2014: A retrospective study based on the national birth defects surveillance system[J]. PLoS One, 2019, 14(6): e0217835. DOI: 10.1371/journal.pone.0217835.
[6] MALTA M, ALMUTIRI R, MARTIN C S, et al. Holoprosencephaly: Review of embryology, clinical phenotypes, etiology and management[J]. Children (Basel), 2023, 10(4): 647. DOI: 10.3390/children10040647.
[7] TAVANO I, DE KEERSMAECHER B, AERTSEN M, et al. Prenatal diagnosis of middle interhemispheric variant of holoprosencephaly: Review of literature and prenatal case series[J]. The Journal of Maternal-fetal & Neonatal Medicine, 2022, 35(25): 4976-4984. DOI: 10.1080/14767058.2021.1873942.
[8] LEWIS A J, SIMON E M, BARKOVICH A J, et al. Middle interhemispheric variant of holoprosencephaly: A distinct cliniconeuroradiologic subtype[J]. Neurology, 2002, 59(12): 1860-1865. DOI: 10.1212/01.wnl.0000037483.31989.b9.
[9] WEISS K, KRUSZKA P, GUILLEN SACOTO M J, et al. In-depth investigations of adolescents and adults with holoprosencephaly identify unique characteristics[J]. Genetics in Medicine, 2018, 20(1): 14-23. DOI: 10.1038/gim.2017.68.
[10] SIMON E M, HEVNER R F, PINTER JD, et al. The middle interhemispheric variant of holoprosencephaly[J]. American Journal of Neuroradiology, 2002, 23(1): 151-156.
[11] 刘俊刚, 李欣, 陈静, 等. 儿童前脑无裂畸形半球中央变异型的影像学诊断[J]. 放射学实践, 2010, 25(12): 1316-1318. DOI: 10.3969/j.issn.1000-0313.2010.12.006. LIU J G, LI X, CHEN J, et al. Imaging diagnosis of pediatric middle interhemispheric variant of holoprosencephaly[J]. Radiologic Practice, 2010, 25(12): 1316-1318. DOI: 10.3969/j.issn.1000-0313.2010.12.006.
[12] öZDEMIR M, TURAN A, KAVAK R P. Middle interhemispheric variant of holoprosencephaly in an asymptomatic adult[J]. BJR Case Reports, 2019, 5(4): 20190035. DOI: 10.1259/bjrcr.20190035.
[13] YAHYAVI-FIROUZ-ABADI N, PORETTI A, IDOWU O R, et al. Case 236: middle interhemispheric variant of holoprosencephaly[J]. Radiology, 2016, 281(3): 969-974. DOI: 10.1148/radiol.2016150503.
[14] BULAKBASI N, CANCURI O, KOCAOğLU M. The middle interhemispheric variant of holoprosencephaly: magnetic resonance and diffusion tensor imaging findings[J]. The British Journal of Radiology, 2016, 89(1063): 20160115. DOI: 10.1259/bjr.20160115.
[15] VOLPE P, CAMPOBASSO G, DE ROBERTIS V, et al. Disorders of prosencephalic development[J]. Prenatal Diagnosis, 2009, 29(4): 340-354. DOI: 10.1002/pd.2208.
[16] MARTINEZ A F, KRUSZKA PS, MUENKE M. Extracephalic manifestations of nonchromosomal, nonsyndromic holoprosencephaly[J]. American journal of medical genetics. Part C, Seminars in Medical Genetics, 2018, 178(2): 246-257. DOI: 10.1002/ajmg.c.31616.
[17] 熊南翔, 赵洪洋, 张方成, 等. 颅骨骨瘤临床分类和手术方法的探讨[J]. 中华神经外科杂志, 2006, 22(3): 166-167. DOI: 10.3760/j.issn:1001-2346.2006.03.013. [18] PATEL T R, BORAH G L. Frontal bone periosteal osteomas[J]. Plastic and Reconstructive Surgery, 2004, 114(3): 648-651. DOI: 10.1097/01.prs.0000130935.83413.6b.
[19] LANGLIE J A, HULLFISH H, JABORI S K, et al. Diagnosis and management of craniofacial osteomas[J]. The Journal of Craniofacial Surgery, 2023, 34(5): 1515-1521. DOI: 10.1097/SCS.0000000000009395.
[20] ATALAR M H, ICAGASIOGLU D, SENER R N. Middle interhemispheric variant of holoprosencephaly associated with bilateral perisylvian polymicrogyria[J]. Pediatrics International, 2008, 50(2): 241-244. DOI: 10.1111/j.1442-200X.2007.02311.x.
[21] 党连荣, 南雅炫, 裴慧杰, 等. 脑裂畸形的CT、MRI影像学征象分析[J]. 国际放射医学核医学杂志, 2014, 38(4): 239-241, 270. DOI: 10.3760/cma.j.issn.1673-4114.2014.04.008. DANG L R, NAN Y X, PEI H J, et al. Analysis of the CT and MRI imaging features of schizencephaly[J]. International Journal of Radiation Medicine and Nuclear Medicine. 2014, 38(4): 239-241, 270. DOI:10.3760/cma.j.issn.1673-4114.2014.04.008. (in Chinese).
[22] 孙凯敏, 李敏, 王伟新, 等. 多层螺旋CT和MRI在额骨内板增生症诊断中的应用价值[J]. CT理论与应用研究, 2021, 30(5): 645-652. DOI: 10.15953/j.1004-4140.2021.30.05.13. SUN K M, LI M, WANG W X, et al. Application value of multi-detector CT and MRI in the diagnosis of hyperostosis frontalis interna[J]. CT Theory and Applications, 2021, 30(5): 645-652. DOI: 10.15953/j.1004-4140.2021.30.05.13.
 下载:
下载:

计量
- 文章访问数: 12
- HTML全文浏览量: 0
- PDF下载量: 4


